Обезвреживание аммиака в организме
В организме человека подвергается распаду около 70 г аминокислот в сутки, при этом в результате реакций дезаминирования и окисления биогенных аминов освобождается большое количество аммиака, являющегося высокотоксичным соединением. Поэтому концентрация аммиака в организме должна сохраняться на низком уровне. Действительно, уровень аммиака в крови в норме не превышает 60 мкмоль/л (это почти в 100 раз меньше концентрации глюкозы в крови). В опытах на кроликах показано, что концентрация аммиака 3 ммоль/л является летальной. Таким образом, аммиак должен подвергаться связыванию в тканях с образованием нетоксичных соединений, легко выделяющихся с мочой.
Один из путей связывания и обезвреживания аммиака в организме, в частности в мозге, сетчатке, почках, печени и мышцах,– это биосинтез глутамина (и, возможно, аспарагина). Глутамин и аспарагин выделяются с мочой в небольшом количестве. Было высказано предположение, что они выполняют скорее транспортную функцию переноса аммиака в нетоксичной форме. Ниже приводится химическая реакция синтеза глутамина, катализируемого глутаминсинтетазой.
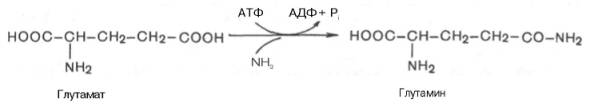
Механизм этой синтетазной реакции, подробно изученный А. Майсте-ром, включает ряд стадий. Синтез глутамина в присутствии глутамин-синтетазы может быть представлен в следующем виде:
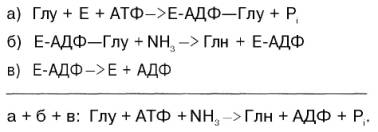
Биосинтез аспарагина протекает несколько отлично и зависит от природы ферментов и донора аммиака. Так, у микроорганизмов и в животных тканях открыта специфическая аммиакзависимая аспарагинсинтетаза, которая катализирует синтез аспарагина в две стадии:
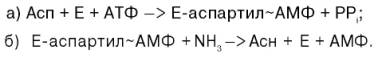
В животных тканях содержится, кроме того, глутаминзависимая аспа-рагинсинтетаза, которая для синтеза во второй стадии использует амидную группу глутамина:
Суммарная ферментативная реакция синтеза аспарагина может быть представлена в следующем виде:
Асп + АТФ + NН3 (или Глн) –> Асн + АМФ + РРi + (Глу).
Видно, что энергетически синтез аспарагина обходится организму дороже, поскольку образовавшийся РРi далее распадается на ортофосфат.
Часть аммиака легко связывается с α-кетоглутаровой кислотой благодаря обратимости глутаматдегидрогеназной реакции. Если учесть связывание одной молекулы аммиака при синтезе глутамина, то нетрудно видеть, что в организме имеется хорошо функционирующая система, связывающая две молекулы аммиака:
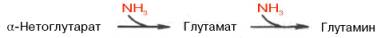
Аммиак в клетках мозга обезвреживается путем
Кафедра поликлинической терапии ФГБОУ ВО МГМСУ им. А.И. Евдокимова Минздрава России, Москва
Основным источником образования аммиака в организме человека является азот пищевого белка, образующийся в ходе реакций дезаминирования аминокислот в печени. Дополнительными источниками образования аммиака являются:
Выделение мочевины осуществляется преимущественно через почки (около 80%), примерно 20% мочевины повторно поступают в ЖКТ, где вновь разлагается уреазаположительными бактериями до аммиака.
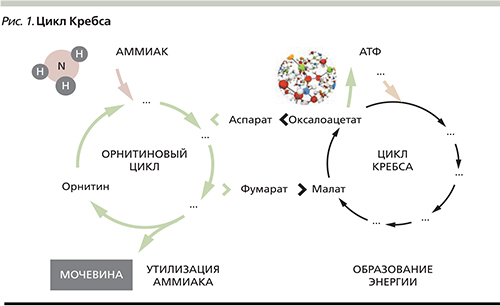
Детоксикация аммиака в организме осуществляется преимущественно в митохондриях перипортальных гепатоцитов за счет связывания в орнитиновом цикле с аминокислотами и образованием нетоксичной мочевины (рис. 1). Частично детоксикация аммиака происходит в мышечной ткани в процессе синтеза глутамина при участии фермента глутаминсинтетазы. Эта реакция с меньшей интенсивностью протекает также в астроцитах головного мозга и перивенозных гепатоцитах печени. Образующийся в результате этих превращений глутамин нетоксичен и выделяется с мочой.
Таким образом, являясь основным источником аммиака, печень в то же время служит главным местом его обезвреживания. Именно поэтому гипераммониемия развивается в организме человека, прежде всего при хронических заболеваниях печени (ХЗП). К причинам этого относятся снижение активности орнитинового цикла и глутаминсинтетазной реакции при печеночно-клеточной недостаточности и порто-системное шунтирование при развитии и прогрессировании портальной гипертензии.
Значительно реже в рутинной клинической практике встречаются генетически детерминированные ферментопатии, которые сопровождаются повышением концентрации аммиака в сыворотке крови. В зависимости от дефицита или дефекта того или иного фермента выделяют несколько видов генетических заболеваний: гипераммониемию типа I (в основе – дефект карбамоилфосфатсинтетазы I), гипераммониемию типа II (в основе – дефект орнитинкарбамоилтрансферазы), цитруллинемию (в основе – дефект аргининосукцинатсинтетазы), аргининосукцинатурию (в основе – дефект аргининосукцинатлиазы), гипераргининемию (в основе – дефицит аргиназы).
Известно, что аммиак является одним из важнейших нейротоксических метаболитов в организме человека. В клинической практике наиболее частым проявлением гипераммониемии является печеночная энцефалопатия (ПЭ) при патологии печени, представляющая собой спектр нервно-психических расстройств на фоне острой или хронической печеночно-клеточной недостаточности и/или портосистемном шунтировании крови. В основе патогенеза ПЭ лежит дисбаланс аминокислот в головном мозге, приводящий к отеку астроглии и нарушениям ее функций, таких как изменения постсинаптических рецепторов и процессов нейротрансмиссии, нарушение проницаемости гематоэнцефалического барьера, снижение энергетического обеспечения нейронов. Степень клинических проявлений напрямую коррелирует с уровнем аммиака в сыворотке крови. В зависимости от выраженности нарушений деятельности головного мозга выделяют четыре степени тяжести печеночной энцефалопатии (от минимальной до комы).
В последние годы в связи с совершенствованием диагностических методик актуально отделение клинически выраженных стадий ПЭ (дезориентация, атаксия, кома) от стадии с минимально выраженными проявлениями (латентная ПЭ). Такую ПЭ можно выявить, используя специальные опросники или метод вызванных потенциалов головного мозга. При этом выявляются когнитивные и психомоторные расстройства, такие как трудности с принятием решений, снижение скорости психомоторных реакций и др. К клиническим симптомам латентной ПЭ относятся повышенная утомляемость, слабость, раздражительность, инверсия сна (сонливость днем и бессонница ночью), нарушения речи, изменения почерка, рассеянность за рулем и при выполнении работы, требующей повышенной концентрации внимания, тремор, снижение мышечных рефлексов.
К сожалению, результатом таких нарушений могут стать серьезные дорожно-транспортные происшествия с тяжелыми последствиями [1, 2]. В связи с этим выявление минимальной ПЭ имеет большое значение для работников многих профессий: водителей автотранспорта, операторов на автоматизированном оборудовании и др.
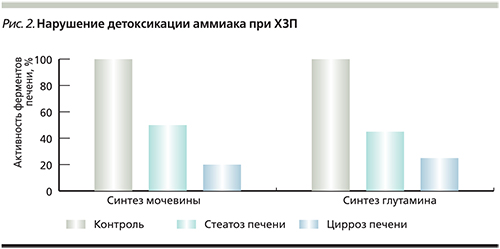
В исследованиях показано, что у больных стеатозом печени при отсутствии клинических признаков воспаления и печеночно-клеточной недостаточности уже имеется выраженное снижение детоксикации аммиака по обоим путям (уменьшается синтез и мочевины, и глутамина) за счет выраженного снижения активности соответствующих ферментов в печени (рис. 2).
В 2016 г. ученые из Великобритании получили новые научные данные, свидетельствующие о том, что гипераммониемия активирует звездчатые клетки печени (ЗКП) и как следствие – может приводить к усиленному коллагенообразованию и прогрессирующему фиброзированию [3].
Известно, что ЗКП являются основными профиброгенными клетками органа. Хроническое повреждение печени под влиянием различных этиологических факторов (алкоголь, вирусная инфекция, лекарства, холестаз и др.) способствует их активации и дифференцировке в миофибробластоподобные клетки, которые приобретают сократительные, провоспалительные и фиброгенетические свойства. При этом ЗКП пролиферируют, из них исчезают капли жира, увеличивается шероховатая эндоплазматическая сеть, в них появляется специфический белок гладких мышц (α-актин), увеличивается количество рецепторов к цитокинам, стимулирующим пролиферацию и фиброгенез.
К факторам, активирующим ЗКП, относятся трансформирующий фактор роста (TGF-β1 – Transforming growth factor beta), тромбоцитарный фактор роста (PDGF – Platelet-derived growth factor), фактор роста фибробластов, интерлейкин-1 (ИЛ-1), эпидермальный фактор роста, фактор некроза опухоли α. Среди всех факторов роста TGF-β1 позиционируется как ключевой медиатор в фиброгенезе у человека. Различные способы, воздействующие на синтез этого фактора или на сигнальные пути, которые реализуются с участием этого фактора, значимо снижают фиброз в экспериментальных моделях [4].
Активированные ЗКП мигрируют и аккумулируются в месте поражения ткани печени, при этом секретируя большое количество внеклеточного матрикса и одновременно регулируя деградацию этих молекул на уровнях транскрипции и посттранскрипции. Повышение содержания информационной коллагеновой РНК является опосредующим фактором, повышающим синтез коллагена активированными ЗКП. В этих клетках посттранскрипционная регуляция коллагена осуществляется путем последовательности 3-го нетранслируемого региона РНК-связующего протеина aСР2, равно как и посредством структуры в 5-м окончании коллагена информационной РНК. Кроме этого ЗКП экспрессируют большое количество нейроэндокринных маркеров (реелин, нестин, нейротрофины, синаптофизин и глиально-фибриллярные кислотные протеины), а также несут рецепторы нейротрансмиттеров, выделяют провоспалительные цитокины, нейрофильный и моноцитарный хемоаттрактаны, которые усиливают воспалительную реакцию в пораженной печени [5].
Фиброз печени является основным, этиологически независимым путем прогрессирования хронических диффузных заболеваний печени вплоть до цирроза. Печеночный фиброз ассоциируется с изменением количества и качественного состава экстрацеллюлярного коллагенового матрикса (ЭКМ). При выраженных стадиях фиброза печень содержит приблизительно в 6 раз больше ЭКМ, чем в норме, а в его составе определяются коллагены (1-го, 3 и 4-го типов), фибронектин, ундулин, эластин, ламинин, гиалуронан и протеогликаны. Снижение скорости резорбции ЭКМ и выведение молекул металлопротеиназ являются в основном следствием перевысвобождения их специфических ингибиторов (TIMPs – tissue inhibitors of metalloproteinases). Результатом превалирования процессов образования внеклеточного матрикса над его разрушением является формирование фиброзного рубца, при этом фиброз на ранних стадиях развития – процесс обратимый, а цирроз с характерными сшивками между коллагеновыми волокнами и узлами регенерации необратим. Прогрессирующее накопление и отложение внеклеточного матрикса в пространстве Диссе приводят к исчезновению фенестров эндотелия, капилляризации и стенозированию синусоидов с постепенным развитием портальной гипертензии [6–8].
Таким образом, в прогрессировании ХЗП главенствующую роль играют повреждения и ишемия гепатоцитов, которые запускают регенераторные процессы воспаления и коллагенообразования. В свою очередь повышенное коллагенообразование из-за избыточного отложения внеклеточного матрикса и нарушения портального кровотока также приводит к ишемии и некрозу гепатоцитов. Таким образом, можно говорить о «circulus vitiosus» (порочном круге) в прогрессировании заболеваний печени.
Почему ученые из Великобритании считают, что гипераммониемия способна выступить важным фактором в этом «circulus vitiosus»? Такие выводы были сделаны на основе результатов собственного исследования, состоявшего из двух частей: in vitro и in vivo. Первичные ЗКП, полученные из печени здоровых доноров (hHSCs – human hematopoietic stem cells), были посеяны (плотность – 26×103/см2) в исходных, богатых сывороткой условиях (CM – полная среда) на 24 часа с последующим удалением сыворотки на следующие 24 часа (SFM – Serum-free medium). Экзогенный глутамин был удален из культуральной среды во избежание искажения результатов эксперимента.
In vitro показано, что аммиак дозозависимо снижает клеточную пролиферацию и метаболизм в первичных человеческих ЗКП, но не вызывает гибели клеток.
Длительная обработка клеток аммиаком (до 72 часов) индуцирует развитие и прогрессирование эндоретикулярного стресса в hHSCs, что проявлялось выраженным перинуклеарным накоплением красителя (ER-Tracker™ Red) и появлением цитоплазматических вакуолей по мере роста концентрации аммиака. Использование Image IT™-набора для определения зеленых активных форм кислорода позволило исследователям выявить аммиак-дозозависимое образование активных радикалов кислорода (ROS – Reactive oxygen species) в hHSCs. Таким образом, установлено, что увеличение концентрации аммиака и время его воздействия напрямую влияют на уровни мРНК экспрессии маркеров стресса [3].
Исследование также показало, что при повышенных концентрациях аммиака hHSCs приобретают выраженный профиброгенный и провоспалительный потенциал, что подтверждается результатами исследования in vitro:
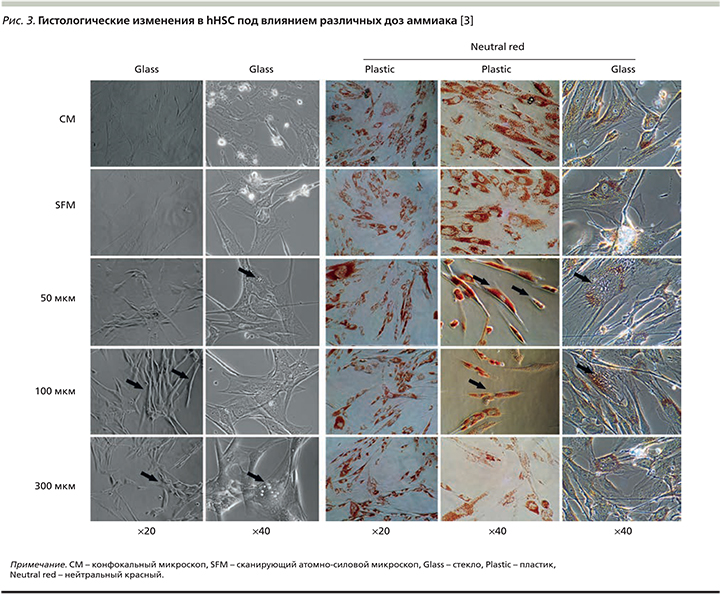
При гистологическом изучении клеток установлено, что аммиак дозозависимо вызывает серьезные морфологические изменения. Так, при световой микроскопии и в тесте жизнеспособности с нейтральным красным (20х, 40х) наблюдали превращение миофибробластоподобных клеток в веретеноподобные фибробласты под действием повышенных концентраций аммиака (рис. 3). Электронная микроскопия показала изменения структуры цитоскелета клеток с образованием цитоплазматических вакуолей при гипераммониемии, однако эти изменения быстро регрессировали после помещения клеток в безаммиачную среду [3].
Вторая часть исследования была проведена на самцах крыс, которые были разделены на три группы: в двух группах проведено перевязывание общего желчного протока (BDL – экспериментальная модель холестаза) с целью формирования экспериментального повреждения печени, при этом крысам одной из групп вводился орнитин (ОР), второй – физиологический раствор. Третья группа животных была контрольной, без повреждения печени.
Анализ результатов опыта показал, что концентрации аммиака были значительно увеличены в плазме крыс с BDL по сравнению с контролем (182±12,8 против 62,51±6,2 мкМ; p
1. Bajaj J.S., Pinkerton S.D., Sanyal A.J., Heuman D.M. Diagnosis and treatment of minimal hepatic encephalopathy to prevent motor vehicle accidents: a cost-effectiveness analysis. Hepatology. 2012;55(4):1164–71.
2. Богомолов П.О., Буеверов А.О., Уварова О.В., Мациевич М.В. Гипераммониемия у пациентов с заболеваниями печени на доцирротической стадии: возможно ли это? Клин. перспективы гастроэнтерол., гепатол. 2013;5:3–8.
3. Джалан Р., Де Чиара Ф., Баласубраманиян В., Андреола Ф., Кхетан В., Малаго М., Пинзани М., Мукерджи Р.П., Ромбоутс К. Аммиак приводит к патологическим изменениям в звездчатых клетках печени, и является целью при лечении портальной гипертензии. Журн. гепатологии. 2016;64:823–33.
4. Gressner A.M., Weiskirchen R., Breitkopf K., Dooley S. Roles of TGF-beta in hepatic fibrosis. Front. Biosci. 2002;7:d793–d807.
5. Lindquist J.N., Parsons C.J., Stefanovic B., Brenner D.A. Regulation of alpha1(I) collagen messenger RNA decay by interactions with alphaCP at the 3?-untranslated region. J. Biol. Chem. 2004;279:23822–29.
6. Benyon R.C., Iredale J.P. Is liver fibrosis reversible? Gut. 2000;46:443–46.
7. Arthur M.J. Fibrogenesis II. Metalloproteinases and their inhibitors in liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2000;279:G245–G249.
8. Arthur M.J. Reversibility of liver fibrosis and cirrhosis following treatment for hepatitis C. Gastroenterology. 2002;122:1525–28.
9. Агеева Е.А., Алексеенко С.А. Опыт применения пероральной формы препарата «L-орнитин-L-аспартат» при гипераммониемии у больных с хроническими заболеваниями печени на доцирротической стадии. Клин. перспективы гастроэнтерологии, гепатологии. 2015;6:24–6.
10. Бурков С.Г., Арутюнов А.Г., Годунова С.А., Гурова Н.Ю., Егорова Н.В., Должикова Т.А., Шиковная Ю.Н. Эффективность гранул L-орнитин-L-аспартата в лечении неалкогольной жировой болезни печени. Consilium Medicum. 2010;12(8):43–7.
11. Осипенко М.Ф., Редькина А.В., Бикбулатова Е.А., Моисеенко Е.Е., Скалинская М.А., Казакова Е.А. Оценка L-орнитин-Lаспартата (Гепа-Мерц) в комплексном лечении неалкогольного стеатогепатита. Consilium Medicum. Прил. Гастроэнтерология. 2010;1:35–8.
12. Грюнграйфф К., Ламберт-Бауманн Й. Эффективность гранул L-орнитин-L-аспартата при лечении хронических заболеваний печени. Сучасна гастро-ентерологія. 2008;2:59–67.
13. Ермолов С.Ю., Шабров А.В., Ермолова Т.В. и др. Новые подходы к диагностике и коррекции портопеченочной гемодинамики. Эксперим. и клин. гастроэнтерология. 2007;4:13–6.
14. Ермолова Т.В., Яковлева Д.М. Эффективность применения L-орнитина-L-аспартата у больных стеатогепатитом. Соврем. гастроэнтерология и гепатология. 2012;1:22–6.
V. Обмен аммиака
А. Источники аммиака в клетках
Таблица 9-3.
Основные источники аммиака


Аммиак — токсичное соединение. Даже небольшое повышение его концентрации оказывает неблагоприятное действие на организм, и прежде всего на ЦНС. Так, повышение концентрации аммиака в мозге до 0,6 ммоль вызывает судороги. К симптомам гипераммониемии относят тремор, нечленораздельную речь, тошноту, рвоту, головокружение, судорожные припадки, потерю сознания. В тяжёлых случаях развивается кома с летальным исходом.
Б. Связывание (обезвреживание) аммиака
Высокая интенсивность процессов дезаминирования аминокислот в тканях и очень низкий уровень аммиака в крови свидетельствуют о том, что в клетках активно происходит связывание аммиака с образованием нетоксичных соединений, которые выводятся из организма с мочой. Эти реакции можно считать реакциями обезвреживания аммиака. В разных тканях и органах обнаружено несколько типов таких реакций.
Основной реакцией связывания аммиака, протекающей во всех тканях организма, является синтез глутамина под действием глутаминсинтетазы:

Глутаминсинтетаза локализована в митохондриях клеток, для работы фермента необходим кофактор — ионы Mg 2+ Глутаминсинтетаза — один из основных регуляторных ферментов обмена аминокислот и аллостерически ингибируется АМФ, глюкозо-6-фосфатом, а также Гли, Ала и Гис.
Глутамин легко транспортируется через клеточные мембраны путём облегчённой диффузии (для глутамата возможен только активный транспорт) и поступает из тканей в кровь. Основными тканями-поставщиками глутамина служат мышцы, мозг и печень. С током крови глутамин транспортируется в кишечник и почки.
В клетках кишечника под действием фермента глутаминазы происходит гидролитическое освобождение амидного азота в виде аммиака:

Образовавшийся в реакции глутамат подвергается трансаминированию с пируватом. а-Аминогруппа глутаминовой кислоты переносится в состав аланина (рис. 9-10). Большие количества аланина поступают из кишечника в кровь воротной вены и поглощаются печенью. Около 5% образовавшегося аммиака удаляется в составе фекалий, небольшая часть через воротную вену попадает в печень, остальные
90% выводятся почками.

Рис. 9-10. Метаболизм азота глутамина в кишечнике
В почках также происходит гидролиз глутамина под действием глутаминазы с образованием аммиака. Этот процесс является одним из механизмов регуляции кислотно-щелочного равновесия в организме и сохранения важнейших катионов для поддержания осмотического давления. Глутаминаза почек значительно индуцируется при ацидозе, образующийся аммиак нейтрализует кислые продукты обмена и в виде аммонийных солей экскретируется с мочой (рис. 9-11). Эта реакция защищает организм от излишней потери ионов Na+ и К+, которые также могут использоваться для выведения анионов и утрачиваться. При алкалозе количество глутаминазы в почках снижается.

Рис. 9-11. Метаболизм амидного азота глутамина в почках
В почках образуется и выводится около 0,5 г солей аммония в сутки.
Высокий уровень глутамина в крови и лёгкость его поступления в клетки обусловливают использование глутамина во многих анаболических процессах. Глутамин — основной донор азота в организме. Амидный азот глутамина используется гпя синтеза пуриновых и пиримидиновых нуклеотидов, аспарагина, аминосаха-ров и других соединений (рис. 9-12).

Рис. 9-12. Пути использования глутамина в организме
Ещё одной реакцией обезвреживания аммиака в тканях можно считать синтез аспарагина под действием аспарагинсинтетазы.
Существуют 2 изоформы этого фермента — глутаминзависимая и аммиакзависимая, которые используют разные доноры амидных групп. Первая функционирует в животных клетках, вторая преобладает в бактериальных клетках, но присутствует и у животных. Однако такой путь обезвреживания аммиака в клетках человека используется редко и к тому же требует больших энергетических затрат (энергию двух макроэргических связей), чем синтез глутамина.
Наиболее значительные количества аммиака обезвреживаются в печени путём синтеза мочевины. В первой реакции процесса аммиак связывается с диоксидом углерода с образованием карба-моилфосфата, при этом затрачиваются 2 молекулы АТФ. Реакция происходит в митохондриях гепатоцитов под действием фермента карбамоилфос-фатсинтетазы I. Карбамоилфосфатсинтетаза II локализована в цитозоле клеток всех тканей и участвует в синтезе пиримидиновых нуклеотидов (см. раздел 10). Карбамоилфосфат затем включается в орнитиновый цикл и используется для синтеза мочевины.
Из мышц и кишечника избыток аммиака выводится преимущественно в виде аланина. Этот механизм необходим, так как активность глутаматдегидрогеназы в мышцах невелика и непрямое дезаминирование аминокислот малоэффективно. Поэтому в мышцах существует ещё один путь выведения азота. Образование аланина в этих органах можно представить следующей схемой (см. схему).
Схема

Аминогруппы разных аминокислот посредством реакций трансаминирования переносятся на пируват, основным источником которого служит процесс окисления глюкозы.
Мышцы выделяют особенно много аланина в силу их большой массы, активного потребления глюкозы при физической работе, а также потому, что часть энергии они получают за счёт распада аминокислот. Образовавшийся аланин поступает в печень, где подвергается непрямому дезаминированию. Выделившийся аммиак обезвреживается, а пируват включается в глюконеогенез. Глюкоза из печени поступает в ткани и там, в процессе гликолиза, опять окисляется до пирувата (рис. 9-13).

Рис. 9-13. Глюкозо-аланиновый цикл
Образование аланина в мышцах, его перенос в печень и перенос глюкозы, синтезированной в печени, обратно в мышцы составляют глюкозо-аланиновый цикл, работа которого сопряжена с работой глюкозо-лакгатного цикла (см. раздел 7).
Совокупность основных процессов обмена аммиака в организме представлена на рис. 9-14. Доминирующими ферментами в обмене аммиака служат глутаматдегидрогеназа и глутаминсинтетаза.

Рис. 9-14. Обмен аммиака. Основной источник аммиака — аминокислоты. Большая часть образовавшегося аммиака обезвреживается в орнитиновом цикле в печени и выделяется в виде мочевины. Основной реакцией обезвреживания аммиака в тканях является синтез глутамина, который затем используется в анаболических процессах и для обезвреживания веществ в печени. Ферменты глутаматдегидрогеназа и глутаминсинтетаза являются регуляторными и обусловливают скорость процессов образования и обезвреживания аммиака
Аммиак в клетках мозга обезвреживается путем
В организме человека подвергается распаду около 70 г аминокислот в сутки; при этом в результате дезаминирования, трансаминирования и окисления биогенных аминов освобождается большое количество аммиака, являющегося высокотоксичным соединением. Поэтому концентрация аммиака в организме должна сохраняться на низком уровне. И, действительно, уровень аммиака в норме в крови не превышает 1-2 мг/л (это почти в 1000 раз меньше концентрации сахара в крови). На кроликах показано, что концентрация аммиака 50 мг/л является летальной. Таким образом, аммиак должен подвергаться связыванию в тканях с образованием нетоксичных соединений, легко выделяемых с мочой.
Синтез амидов требует доставки энергии в виде АТФ и присутствия глутаминовой или аспарагиновой кислоты, свободного аммиака и катализируется специфическими глутамин- и аспарагинсинтетазами в соответствии с уравнением реакций:

Суммарная ферментативная реакция синтеза аспарагина может быть представлена в следующем виде:
Часть аммиака легко связывается с α-кетоглутаровой кислотой благодаря обратимости глутаматдегидрогеназной реакции; если еще учесть синтез глутамина, то нетрудно видеть, что в организме имеется хорошо функционирующая система, связывающая две молекулы аммиака:

Орнитиновый цикл мочевинообразования
Для объяснения механизма образования мочевины было предложено множество теорий. Одной из них является разработанная М. В. Ненцким схема синтеза мочевины, основанная на допущении, что непосредственными источниками углерода и азота в молекуле мочевины являются аммиак и углекислота (в форме угольной кислоты). Это положение подтвердилось позже, хотя в связи с открытием аргиназы механизм синтеза мочевины оказался другим.
Кребс и Гензелейт в 1932 г. в опытах со срезами печени измеряли образование мочевины при добавлении в среду различных аминокислот и аммонийных солей. Было показано, что добавление к срезам печени аммонийных солей и каталитических количеств орнитина (но не какой-либо другой аминокислоты) приводило к образованию значительно большего количества мочевины, чем стехиометрическое его количество (одна молекула орнитина способствовала синтезу 20 молекул мочевины; поскольку источником азота мочевины были ионы аммония, орнитин действительно оказывал каталитическое действие). К этому времени уже была открыта в печени аргиназа, которая катализировала распад аргинина на орнитин и мочевину: аргинин-oрнитин + мочевина. Кребс полагал, что каталитическую роль орнитина можно было бы объяснить, если бы существовал некий механизм для регенерации аргинина из орнитина, согласно уравнению:

Это предположение стимулировало поиск возможных промежуточных продуктов между орнитином и аргинином и в качестве кандидата была теоретически предсказана аминокислота цитруллин, который до этого был изолирован из столового арбуза (Citrullus). И действительно, в опытах на срезах печени цитруллин оказывал такой же каталитический эффект в присутствии аммонийных солей на выход мочевины, как и орнитин. На основании этих данных. Кребс вывел уравнения реакций синтеза мочевины, которые представлены ниже в виде цикла, получившего в литературе название орнитинового цикла мочевинообразования Кребса (рис. 102).
Следует указать, что в биохимии это была первая циклическая система метаболизма, почти на 5 лет опередившая открытие Кребсом другого метаболического цикла трикарбоновых кислот.
Дальнейшие исследования в основном подтвердили циклический механизм биосинтеза мочевины в печени, однако благодаря исследованиям Коена и Ратнер были уточнены промежуточные этапы, природа других участников и ферментные системы, катализирующие образование мочевины.
Таким образом, весь цикл мочевинообразования может быть более детально представлен следующими уравнениями реакций. На первом этапе синтезируется высокоэртическое соединение карбамоилфосфат из СO2 и NH3 (или глутамина в качестве донатора аммиака); этот синтез требует участия двух молекул АТФ:

Фермент содержит биотин в качестве кофермента и сначала реагирует с одной молекулой АТФ, образуя комплекс активная СO2-Е, который затем реагирует с молекулой аммиака (или амидной группой глутамина) и со второй молекулой АТФ (в качестве донатора фосфата). N-Ацетилглутамат является специфическим активатором реакции и его роль сводится, очевидно, к стабилизации активной формы фермента.


Необходимо учесть, что аргиназа содержится в печени тех животных, которые экскретируют мочевину как основной и конечный продукт азотистого обмена с мочой; в печени птиц, например, аргиназа отсутствует, поскольку птицы выделяют мочевую кислоту вместо мочевины. Таким образом, орнитиновый цикл мочевинообразования с учетом новых данных может быть представлен в следующем виде (рис. 103).
Ниже приведена, кроме того, суммарная реакция синтеза мочевины без учета промежуточных продуктов:

Поскольку имеет место снижение свободной энергии, процесс всегда протекает в направлении синтеза мочевины.
Из представленных выше уравнений нетрудно видеть, что один из атомов мочевины имеет своим источником свободный аммиак или амидную группу глутамина (через карбамоилфосфат); второй атом азота мочевины имеет своим источником аспарагиновую кислоту, а не свободный аммиак, образующийся главным образом в процессе глутаматдегидрогеназной реакции. Что касается пополнения запасов аспарагиновой кислоты, то в этом процессе участвуют три сопряженные реакции: сначала фумаровая кислота под действием фумаразы, катализирующая присоединение молекулы Н2O, превращается в яблочную кислоту, которая подвергается окислению при участии специфической малатдегидрогеназы с образованием щавелевоуксусной кислоты; последняя трансаминируется с глутаминовой кислотой, давая аспарагиновую кислоту.
В процессе эволюции живые организмы выработали различные типы азотистого обмена. Аммонийтелический тип, когда главным конечным продуктом азотистого обмена служит аммиак, свойственен преимущественно рыбам. Уротелический тип обмена, когда основным конечным продуктом обмена белков является мочевина, характерен для человека и животных. И, наконец, урикотелический тип, когда главным конечным продуктом обмена является мочевая кислота, имеет место у птиц и рептилий.



